


We were pleased to be able to provide partial support for three summer scholars in the SENS Research Foundation 2015 Summer Scholars Program.
- Celine-Lea Halioua-Haubold, SRF Summer Scholar at the Sanford-Burnham Medical Research Institute
Celine studied a non-toxic subunit of cholera toxin, cholera toxin B (CTB), as a way to introduce therapeutics into glioblastoma cells. Her work suggested that CTB may be an effective way to deliver therapy to glioblastoma. Read Celine's summary of her work here.
- Neal Nathan, SRF Summer Scholar also at the Sanford-Burnham Medical Research Institute
Neal used human induced pluripotent stem cells to study transcription factor EB (TFEB). He compared cells derived from Parkinson's Disease patients and normal controls. His work made a valuable contribution to knowledge about how TFEB may be used to treat Parkinson's. Read Neal's summary of his work here.
- Le Zhang, SRF Summer Scholar at the Scripps Research Institute
Le did important work to find a methodology to show the genetic stability of differentiated iPS cells. He completed painstaking gene by gene analysis of cell clones to look for deletions, duplications, loss of heterogenicity, copy number variation, and the presence or absence of tumerogenic genes. Later, he used computer analysis of SNP data from clones to show the temporal stability of differentiated cells. This work is crucial to complete before these differentiated cells can be used for therapy. Le's work is a vital step forward.
We are amazed at the accomplishments of these three scientists. We are very pleased to be able to post their profiles:
Celine-Lea Halioua-Haubold

My name is Celine-Lea Halioua-Haubold, and I am a rising junior at the University of Texas at Austin where I am double majoring in Chemistry and Neuroscience. I was immediately captivated by neuroscience when shadowing a radiologist, where I was able for the first time to see the inner workings of the body. My interest in chemistry developed due to my placement in a lab studying nanomaterials by my university's Freshman Research Initiative, an area which initially intimidated but now fascinates me.
The majority of my research experience has come from working under Regina Mangieri, Ph.D., and Tira Meyer, Pharm.D., members of Dr. Richard Morrisett's neuropharmacology lab. The lab hopes to uncover the neuronal changes resulting from prolonged alcohol exposure by modeling the development of addiction in mice models. I assisted Dr. Meyer in conditioning her mice to drink ethanol along with basic mouse care. I also assisted Dr. Mangieri in her project to verify the effectiveness of Cholera Toxin subunit-B conjugated to Alexa Fluor-555 (CTB) as a fluorescent retrograde tracer of neuronal pathways. This was of interest due to the theory that different neuronal subtypes are modified differentially by long-term ethanol exposure. It is crucial to be able to differentiate subtypes visually in order to study the changes associated with each one. We conducted surgery to inject the CTB into an area of the brain that is part of the reward pathway in order to label these specific neurons for further experiments. We found that the CTB did succeed in labeling these neurons exclusively.
In Dr. Keith Stevenson and Dr. Stacia Rodenbusch's Nanomaterials for Chemical Catalysis lab, I worked with DNA-templated silver nanoclusters (Ag-NC), a unique fluorescent nanoparticle, which is being investigated for clinical and research uses, such as identifying certain strands of DNA and delivery of drugs. The fluorescing wavelength of these nanoclusters is theoretically completely modifiable depending on the nucleotide sequence of the DNA strand, synthesis pH, and ratio of silver to DNA to reducing agent. Hoping to develop a new neuronal tracing method that is inexpensive, easy to produce, and able to be targeted to specific cell types, I synthesized and injected the Ag-NCs in vivo and then analyzed the distribution and labeling of the neuronal cell bodies. Full analysis of the brain slices has not been completed yet, however it has been provisionally noted that the Ag-NCs were non-toxic, stable in vivo for an extended period of time, and successfully labeled cell bodies. The next steps in the project will be to develop the ability to target them and explore drug delivery methods using the Ag-NCs.
As part of the SRF Summer Scholars Program, I have joined Dr. Evan Snyder's lab of the Sanford Consortium for Regenerative Medicine at the Sanford-Burnham Medical Research Institute. Among his many research interests, Dr. Snyder helped establish the field of neural stem cells and is interested in their potential to repair damaged brain tissue.
Dr. Snyder's lab works in the field known broadly as gene therapy. Gene therapy could allow specialized, highly targeted treatment of a disease by directly remedying the individual afflicted cells. This approach is applicable to most diseases but especially for those where, thus far, treatments have proven ineffective or lacking altogether. The most aggressive of the malignant brain tumors, glioblastoma kills 90% of patients within 3 years even with equally aggressive treatment. There is a desperate need for a treatment that is effective and safe. By understanding the intracellular mechanisms behind the cancer cell's malignancy, it is possible to begin searching for therapies that directly target these pathways to render them inert. This method of treatment is incredibly interesting due to its potential specificity. With a well-targeted treatment, there should be little or no loss of healthy brain cells because the therapeutic targets are not present in healthy cells. This type of gene therapy could be just as deadly to cells as our current methods of cancer treatment - but, most importantly, have specificity to only those cells that are already unhealthy.
Neural stem cells (NSCs) have been shown to exhibit pathotropism: with no modification, they naturally gather in diseased or injured brain tissue. Like all cells, neural stem cells release exosomes. After being released from the parent cells, these nanoscale vesicles, which can carry RNA, DNA, and proteins, travel to their target cell where the vesicle is absorbed and the contents are released into the target cell. NSCs gather in diseased tissue, where they release these exosomes, which in turn enter the neighboring, diseased cells. This phenomenon presents a potential endogenous method of gene therapy delivery. It is theorized that loading the NSCs with a protein or RNA that disables a crucial pathway of the glioblastoma cells would ablate the tumor.
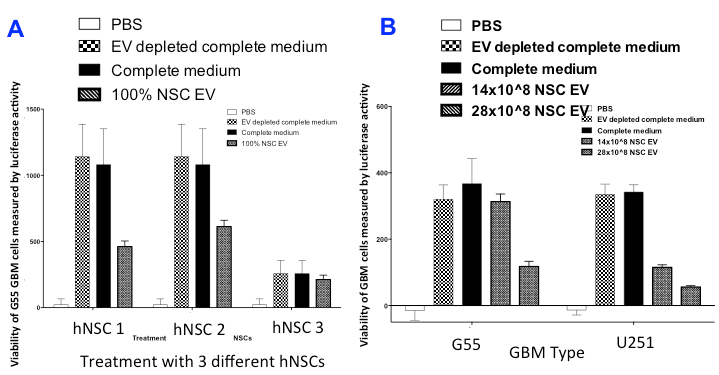
Figure 1. Viability of glioblastoma cells in neural stem cell exosome-rich media Measure of luciferase activity (an artificially introduced luminescent protein) of glioblastoma multiforme (GBM) cells allows a way to visualize changes in cell activity via variations in bioluminescence expression. (A) - comparison of GBM viability in four different cell mediums. There is a distinct lack of viability in the neural stem cell exosome (NSC EV) treated GBMs in comparison to GBM cells in two different mediums. The GBM cells in phosphate buffered saline (PBS) were effectively extinguished due to PBS' inability to support cell growth. (B) - comparison of two different types of GBM cells in their response to NSC EVs. Both cell types showed less viability in the medium with exosomes, with an even further decrease in cell proliferation seen with higher concentration of NSC EVs.
This summer, I will assist Anthony Orona, a graduate student in the Snyder lab, in studying and improving the viability of this novel treatment method. We will develop a protocol to maximize uptake of NSC exosomes by glioblastoma cells in vitro. In order to load these exosomes, we will use viral transfection methods to overexpress various forms of RNA in the NSCs. These varying types of RNA have the ability to prevent the full production of a protein by inhibiting the process at different stages, depending on the characteristics of the RNA. The hope is that the exosomes released by the NSCs will be loaded with these special RNA, enter the glioblastoma cells, and either induce apoptosis or disable further division by strategically blocking certain proteins.
On the side, I will be working on a potential new method of introducing therapeutic materials into cells. Many non-viral methods of transfecting cells do not have a high percentage of efficiency due to the tendency of some cells to deflect the transfection. I will be hijacking a bacterial method of toxin entry into cells for delivery of my desired agents. It will be very important to validate the effectiveness, biosafety, dosing levels, and consistency of the method. I will optimize the method first by transfecting GFP (green fluorescent protein). Should those experiments prove successful, I will attempt to transfect therapeutic proteins into diseased cells. If the method works successfully in vitro by the end of the summer, the efficacy of this method will be tested in a mice model.
Future Plans: I hope to attend a research-intensive medical school where I will be able to continue exploring my research interests as well as train to be a physician. Long term, I hope to find a position in academic medicine, where I may continue to be involved in a lab, work with patients, and help train future generations of doctors. I believe the skill set taught during research is incredibly important for every future physician, even those not planning on actively working in a wet lab, I and hope that I can bring an interdisciplinary research-based approach to teaching medicine as a faculty member.
Celine's Work Summary, Summer 2015
I investigated cholera subunit-B as a method of delivering therapeutics to the quickest and deadliest type of brain cancer, glioblastoma.1 Cholera Toxin Subunit B (CTB) is the protein which is part of the toxin released by V. cholerae. Infections of patients with this bacterium manifests as the disease cholera. CTB itself is non-toxic and interesting to researchers due to its neuronal specificity and efficacy in entering cells. 2,3 I have provisionally shown that by attaching a specific protein to CTB, I could sensitize these cancer cells to a secondary treatment, causing earlier cell death (Figure 1). Data collected also suggested that CTB enters the cell very quickly and does not degrade nor get pushed out of the cell - a fate that befalls many cell-targeting treatments due to the cell’s natural protective measures. Although these pilot experiments will require further study, the analysis suggests the potential of CTB as a therapeutic delivery method.
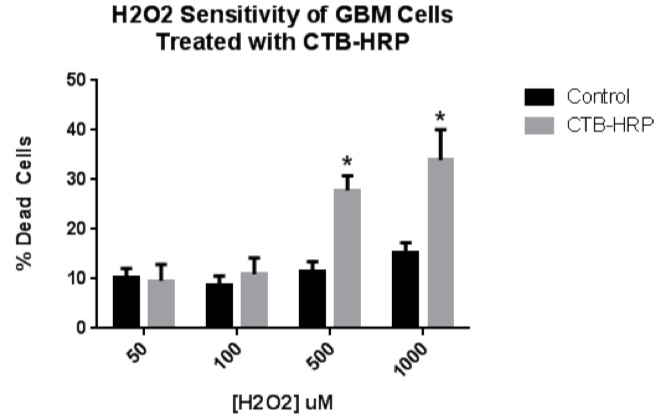
Figure 1. H2O2 sensitivity of CTB-HRP-treated GBM cells. To investigate if a CTB-delivered protein could sensitize GBM cells to a secondary treatment, GBM cells were incubated with CTB-HRP and then exposed to oxidative stress. Cells were incubated with 2 ug/mL of CTB-HRP and then treated with either 50, 100, 500, or 1000 uM hydrogen peroxide diluted into serum-free medium. Cell viability was assessed in experimental and control wells after 24 hours. No statistical difference was observed between the 50 and 100 uM treatments and the control. However, the 500 and 1000 uM treatments produced a statistically-significant higher level of cell death in response to the peroxide treatment.
References:
- Bleeker, Fonnet E.; Molenaar, Remco J.; Leenstra, Sieger. Recent advances in the molecular understanding of glioblastoma". Journal of Neuro-Oncology. 2012;108(1):11–27.
- Bharati, K., & Ganguly, N. K. Cholera toxin : A paradigm of a multifunctional protein Indian J. Res. Med. 2016;133(2):179–187.
- Harris, J. B., Larocque, R. C., Qadri, F., Ryan, E. T., & Calderwood, S. B. Cholera. Lancet. 2016;379:2466–2476.
Neal Nathan

Hello, I am a rising junior at the University of California, Berkeley planning on double-majoring in Molecular and Cell Biology (with an emphasis in Neurobiology) and Public Health and minoring in Public Policy. With an interest in science primarily instilled by my family and early educators and a passion for neurobiology cultivated by the amazingly comedic book Blood, Bones, and Body Bits and competing in Science Olympiad, I have participated in several research experiences studying the nervous system.
During my senior year of high school and summer before college, I interned in the Gage lab at the Salk Institute where I helped a postdoctoral member of the lab, Dr. Jerome Mertens, study the physiological differences between old and young neurons by constructing viral vectors for cellular visualization. Due to previous advances in reprogramming fibroblasts to induced pluripotent stem cells, the lab had a suitable supply of in vitro cellular models of neurons. Since other studies at the Salk Institute showed that age-dependent deterioration of nuclear pore complexes cause reduced nuclear integrity in post-mitotic cells, we sought to explore whether neurons acquired from old and young reprogrammed fibroblast sources exhibit differences in nuclear leakiness. As neurons are quiescent and remain in G0 without undergoing mitosis, they are inherently post-mitotic cells derived from neuroblasts. Consequently, neuronal nuclear pore complexes do not have the benefit of mitosis to regenerate, so they are vulnerable to oxidative stress and accumulated damage.
To characterize this phenomenon in neurons and provide an in vitro model for aged neurons to study age-associated diseases, such as Alzheimer's and Parkinson's, we visualized the different locations of proteins in the cell. To do this, I constructed plasmids that enabled the detection of proteins located in either the cytoplasm or the nucleus, as a colored light would be emitted upon binding to the protein of interest and would be visible via microscope. Following viral transduction of neurons, I viewed cultures and captured overlay pictures using fluorescent microscopy to observe nuclear leakiness. Thus, we could demonstrate that the nucleus was leaky if we saw proteins in the cytoplasm that were supposed to be located primarily in the nucleus or if we saw proteins in the nucleus that were supposed to be located primarily in the cytoplasm. I also visually evaluated neuronal nuclear integrity and form to further describe age-associated alteration to nuclear structure and functionality. From all of this information, the concept of age-associated neuronal leakiness was confirmed. Neuronal nuclear leakiness and malformation were more evident in older neurons than younger neurons. Additional experimentation could verify the statistical significance of these observations and could help explain the pathological processes of age-associated neurodegenerative diseases.
In an additional opportunity to illuminate the underlying causes of neurological conditions, I worked with Dr. Noopur Amin, a postdoctoral member of the Kaufer lab in the UC Berkeley Helen Wills Neuroscience Institute and Department of Integrative Biology, on an autism and depression project that studied the neural circuitry of stress-linked prosocial behavior in rats. Utilizing a rat behavioral paradigm where a free rat learns to help a restrained rat, a product of benevolent behavior, we manipulated neural systems associated with emotion, empathy, movement, and reward through three different methods to explore the neuroscience of altruistic behavior. Optogenetics involves the activation of neurons by shining light through a surgically inserted fiber optic cable in the brain of a rat whose neurons were infected with a virus that enables light reactivity. Adrenalectomies is a surgical method in which the adrenal glands are removed. And, RNA interference is a technique which blocks translation to prevent protein formation. Prosocial behavior is thought to involve numerous neural networks and structures, such as the HPA-axis, the amygdala, and the nucleus accumbens, which respectively implicate a stress response, emotions, and motivation. We looked at these three main pathways and regions, since people with autism or depression exhibit reduced prosocial behavior.
To motivate freeing the restrained rat, I decreased the degradation of acetylcholine in the second rat by using RNA interference to down-regulate acetylcholinesterase activity and stimulated select brain regions through optogenetics. We then visualized brain tissue to observe the implicated neural circuits and assess the consequences of viral, optogenetic, and surgical intervention. Preliminary data indicates that higher corticosterone (a stress hormone) levels and greater activity in the nucleus accumbens and anterior cingulate cortex, which are respectively related to motivation and sense of self, facilitates prosocial behavior. These findings not only provide a better understanding of the physiological characteristics associated with autism and depression but also provide avenues for exploring possible interventions to helping people enduring these conditions.
Wanting to refocus on research related to neurodegenerative diseases, I was led to the SENS Research Foundation with whom I share a vital commitment to combat age-associated disease.
At the Snyder lab of the Sanford Burnham Medical Research Institute and the Sanford Consortium for Regenerative Medicine, I work on a project guided by Dr. Lina Mastrangelo, a postdoctoral member of the lab, that is evaluating the interplay between two proteins implicated in the pathology of Parkinson's Disease (PD). An age-related neurodegenerative disorder, as 60 years is the average age at which people are diagnosed, PD affects one in 100 over the age of 60.1 Mutations to the genes encoding the proteins Alpha-Synuclein, a very common cytosolic neuronal protein, and Leucine-Rich Repeating Kinase 2 (LRRK2), a protein that interacts and influences other through phosphorylation and organelle interactions, are the only known mutations to cause clinical phenotypes closely resembling sporadic cases.2 These mutations lead to accumulation of alpha-synuclein in dopaminergic neurons of the substania nigra, a part of the midbrain involved in movement, and the consequent formation of intracellular protein aggregates called Lewy bodies that are responsible for neuronal cell death and for the clinical outcomes of PD. Impairment of autophagy, which is a way to clean up, breakdown, and export excess proteins from the neurons, may be the main cause of neurodegenerative diseases. Lysosomes are cell organelles capable of breaking down proteins, however the aging process may decrease this ability. In PD, studies suggest the abnormal function of two main protein degradation pathways - macroautophagy and a form of lysososomal degradation called chaperone-mediated autophagy (CMA).3
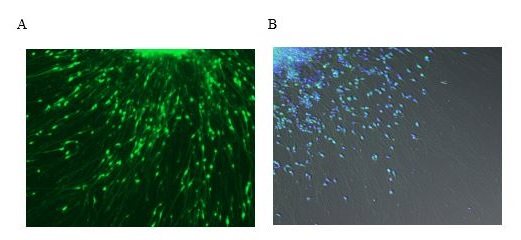
Figure 1. Representative images of alpha-synuclein localization in control neurons Pictured are images of cultured control neurons that were differentiated from iPS cells derived from skin biopsies of unaffected donors. (A) Neurons were transduced with a virus that emits green fluorescence when bound to alpha-synuclein (B) Long exposure imaged control neurons: alpha-synuclein (green) and nuclear DAPI-staining (blue).
The goal of my project is to investigate the mechanisms by which alpha-synuclein and LRRK2 interfere with protein degradation in neurons generated from Parkinson's patients using induced pluripotent stem (iPS) cell techniques. iPS cells are adult cells that are usually obtained from skin biopsies and genetically reprogrammed to resemble embryonic stem cells. Stem cells can grow into any cell type of the body. Thus, to investigate the role of LRRK2 and alpha-synuclein in lysosomal pathways, this technique will be used to generate neuronal cells from PD patients carrying the LRRK2 mutation, patients with a triplication of alpha-synuclein (PARK1 mutation), and unaffected control patients. I will evaluate how LRRK2 and alpha-synuclein influence autophagy by using Western blotting to measure biomarkers for autophagy and by conducting fluorescent microscopy staining to study the localization of key proteins, such as alpha-synuclein. I hope my project will provide insight into the cellular mechanics of Parkinson's disease and improve the development of better therapeutics.
Future Plans: I plan to graduate from UC Berkeley in May 2017 and then pursue a medical degree to become a neurologist. Due to my passions for science and law, I hope to continue to contribute to clinical research and also earn a Masters Degree in Public Health. This way I can help both the patients I see and those I will not see by developing therapeutics for neurodegenerative diseases and implementing progressive healthcare policy.
Neal's Work Summary, Summer 2015
Parkinson's Disease (PD) is the major motor neurodegenerative condition worldwide. PD is an age-related disorder, as the majority of cases are sporadic, and 60 years is the average age at which people are diagnosed. Moreover, PD affects one in 100 over the age of 60 with an estimated one million patients in the United States alone.1 However, genetic mutations associated with early onset of the disease have been identified for about 10 percent of Parkinson’s cases. PD is characterized by extensive loss of dopaminergic neurons in the substantia nigra pars compacta.2 A common feature of PD is the misfolding and aggregation of the protein alpha-synuclein (a-syn) which forms cytoplasmic inclusions in neuronal cells called Lewy bodies.4 In familial PD, the mutated leucine-rich repeating kinase 2 (LRRK2) and a-syn have been linked to impairment in autophagy, which is a biological process responsible for cleaning up, breaking down, and exporting excess proteins from neurons.3 Recent PD studies suggest a neuroprotective role for the transcription factor EB (TFEB) in which it stimulates autophagic lysosomal pathways.5 In this study, we used human induced pluripotent stem cells (hiPSCs) derived from PD patients with a G2019S LRRK2 mutation, patients with a PARK1 mutation, and healthy subjects (controls) to investigate the expression of TFEB. To validate expression of a-syn in the neuronal cells, transduction with a lentivirus targeting a-syn was performed. Western blotting techniques revealed expression of proteins associated with autophagy and PD. Natural amounts of TFEB in PD and control cell lines was measured via Western blot. Also, Human Embryonic Kidney (HEK) 293FT cells were transfected with a TFEB plasmid. Our preliminary data suggest that studies performed on hiPSCs derived from familial PD patients may provide insight into the cellular mechanisms of the disease and potentially support the development of therapeutic treatments targeting TFEB, as the endogenous expression of TFEB is consistent between control and PD cell lines, making the overexpression of TFEB a possible intervention.
References:
- The Michael J. Fox Foundation. "Parkinson's Disease Causes - Parkinson's Disease Information." N.p., n.d. Web. 10 June 2015.
- Liu, Guoxiang, Leonardo Aliaga, and Huaibin Cai. "Α-Synuclein, LRRK2 and Their Interplay in Parkinson’s Disease." Future neurology 7.2 (2012): 145–153. Print.
- Cuervo, Ana Maria, and Esther Wong. "Chaperone-mediated Autophagy: Roles in Disease and Aging." Cell Research 24.1 (2014): 92-104. Nature. Web. 24 May 2015.
- Menzies, Fiona M., Angeleen Fleming, and David C. Rubinsztein. "Compromised Autophagy and Neurodegenerative Diseases." Nature Reviews Neuroscience Nat Rev Neurosci 16.6 (2015): 345-57. Web. 3 July 2015.
- Decressac, Mickael, and Anders Björklund. "Tfeb." Autophagy 9.8 (2013): 1244-246. Web. 19 June 2015.
Le Zhang

My name is Le Zhang. I just completed my Bachelor of Science in biochemistry & biotechnology from Michigan State University. Before joining to the SRF Summer Scholars Program, I worked in Dr. Yonghui Zheng’s lab at MSU searching for novel proteins or molecules from our human body that can limit human immunodeficiency virus-1 (HIV-1) infection. These proteins are called restriction factors. One of the potential restriction factors we researched was the T-cell immunoglobulin and mucin domain (TIM) protein family. TIMs are T cell surface receptors which were reported in the past decade to promote envelope viruses (viruses surrounded by a lipid bilayer) binding and entry into the host cells like hepatitis A and Ebola. 1 However, one of the most recent studies has shown that TIM proteins 1, 3, and 4 s trongly inhibit the release of HIV-1,2 but the study did not establish the mechanism by which viral proteins antagonize TIM proteins.
Under the supervision of Dr. Yonghui Zheng and Dr. Xianfeng Zhang, I focused on verifying if TIMs have the novel ability to inhibit HIV-1 and finding possible viral proteins that interact with TIMs. Our hypothesis was that HIV Negative Regulatory Factor (Nef), a viral protein which can down regulate host surface receptors like CD4, has the ability to also interact with TIMs. To test this hypothesis, I created an HIV-1 Nef knockout mutant in a retro-vector, which is a plasmid that encodes all HIV-1 proteins except for Nef and can assemble HIV particles. Three TIM plasmids were also prepared, which overexpressed TIM 1, 3 & 4 proteins, respectively. I co-transfected HIV-1 Nef knockout and TIMs plasmids in 293T cells, which are specially designed cells to overexpress plasmids. I determined the effect of overexpression of TIMs by measuring the amount of virus released by the Nef knockout strain compared to the non-mutant control. This research helped us better understand how the HIV infection mechanism works and how our immune system responds to the infection. It will serve as the foundation for future anti-viral treatment development in the Zheng lab.
I was attracted to stem cell research after learning about their power in tissue renewal, their utility in fighting aging, and other novel functions. I hope to learn more about stem cells during my internship with the SRF Summer Scholars Program.
This summer, I will be conducting my research project in Dr. Jeanne Loring’s laboratory at the Center for Regenerative Medicine in the Scripps Research Institute. Guided by Dr. Michael Boland and Dr. Andres Bratt-Leal, my main goal for the summer is to analyze the genomic stability of induced pluripotent stem cells (iPSCs) and neuronal progenitor cells (NPCs), which are intended for cell replacement therapies for patients with Parkinson’s disease.
Human iPSCs are usually derived from somatic cells, like skin cells. By using biological or chemical methods, they are reprogrammed into stem cells. iPSCs are well known for their ability to self-renew and their power to differentiate into other cell types. These features make them a promising tool in cell transplantation therapy development. In many age-related diseases, irreversible cell death is often a major contributor. For instance, death of dopamine-generating cells, one type of neuron in the midbrain, leads to Parkinson’s disease. Previous research in rodent and primate models of Parkinson’s disease has shown that midbrain dopaminergic (DA) neurons differentiated from human embryotic stem (ES) cells improved forelimb use and movement control in Parkinsonian animals. 3 Also, ES cell-derived DA neurons showed long-term survival in animals. 3
Use of human ES cells raises some morality concerns and can lead to resistance for use in adult treatments. However, use of iPSCs directly from patients addresses this concern and is more convenient and safer as well. Although iPSC research shows great promise, reprogramming has the potential to induce detrimental genomic changes. Expansion and differentiation of iPSCs over time could lead to genomic change and possibly tumorigenesis.4 Past experiments from the Loring lab indicate that cell lines propagated over 100 continuous passages, regardless of passage methodology, experience genomic instability, such as genomic deletions and duplications. 5
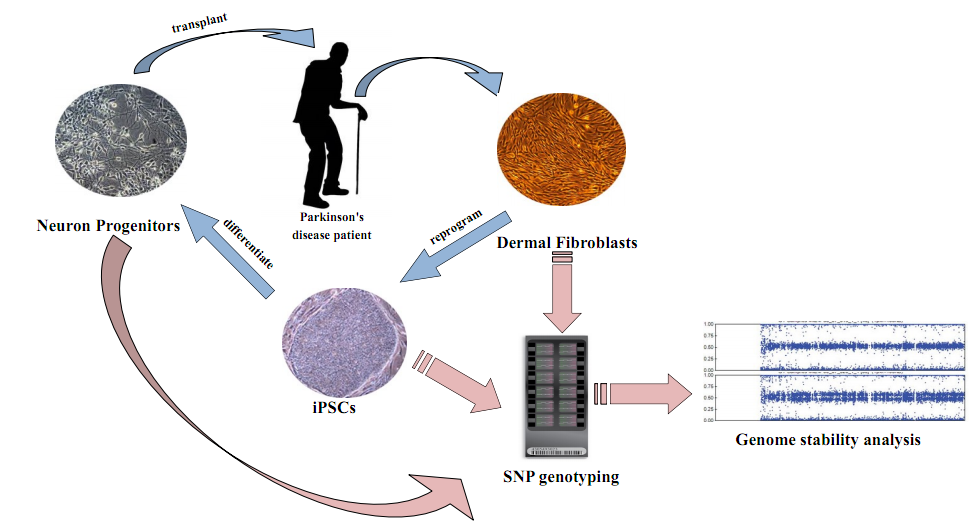
Figure 1. Parkinson's disease transplantation project overview Dermal Fibroblast cells are taken from patients. They are reprogrammed into iPSCs, then differentiated into NPCs, and finally, transplanted back into patients. NPCs will later develop into DA neurons inside patients. I will use SNP genotyping and computational programs to analyze cells from each of the steps and help to select untumorigenic cell lines. From Illumina, Inc. (Laurent, et al, 2011) 6
The Loring lab has derived dermal fibroblasts from 10 patients with Parkinson’s disease. These fibroblasts have been reprogrammed to iPSCs, which have been differentiated into midbrain-specific NPCs. These cells will later develop into DA neurons after transplantation. The Loring lab is the first lab conducting iPSC transplantation on Parkinson’s disease patients, so it is essential to ensure genomic stability of the cells being transplanted. An important method to determine genomic integrity of patients’ iPSC lines is single nucleotide polymorphism (SNP) genotyping, which can be used to examine millions of single base pair differences at genomic sites specific to humans.
SNP analysis will enable me to determine if the cell populations are suitable for transplantation or whether they have too much genetic change and, hence, potential risk for tumorigenesis. My research this summer will generate and analyze genomic SNP profiles from patient-specific dermal fibroblasts, iPSCs, and neuronal progenitors. SNP patterns from the three cell types will be compared to determine whether genomic instability has occurred from fibroblasts to iPSCs then to neuronal progenitors. Hopefully, with efforts from other scientists and me, the Loring Lab will successfully identify some cell lines that are suitable for transplantation and pass the FDA approval.
Future Plans: Stem cell research is new to me. Through my experiences in the Loring lab, I want to learn more and later contribute to this field. I plan to apply to graduate school after the SRF Summer Scholars Program has ended.
References:
- Stephanie Jemielity, Jinyize J Wang, Ying Kai Chan, Asim A Ahmed, Wenhui Li, Sheena Monahan, Xia Bu, Michael Farzan, Gordon J Freeman, Dale T Umetsu, Rosemarie H Dekruyff, Hyeryun Choe. TIM-family Proteins Promote Infection of Multiple Enveloped Viruses through Virion-associated Phosphatidylserine. PLOS. Pathogens: 2013. e1003232. back to text
- Minghua Lia, Sherimay D. Ablanb, Chunhui Miaoa, Yi-Min Zhenga, Matthew S. Fullera, Paul D. Rennertc,Wendy Mauryd, Marc C. Johnsona, Eric O. Freedb,Shan-Lu Liua. TIM-family proteins inhibit HIV-1 release. PNAS: 2014, doi: 10.1073/pnas.1404851111 back to text
- Sonja Kriks, Jae-Won Shim, Jinghua Piao, Yosif M. Ganat, Dustin R. Wakeman, Zhong Xie, Luis Carrillo-Reid, Gordon Auyeung, Chris Antonacci, Amanda Buch, Lichuan Yang, M. Flint Beal, D. James Surmeier, Jeffrey H. Kordower, Viviane Tabar & Lorenz Studer. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature. 2011. Vol 480, 547-551. back to text
- Fox JL FDA scrutinizes human stem cell therapies. Nat Biotechnol. 2008. Vol 26, 598-599. back to text
- Ibon Garitaonandia, Hadar Amir, Francesca Sesillo Boscolo, Geral K. Wambua, Heather L. Schultheisz, Karen Sabatini, Robert Morey, Shannon Waltz, Yu-Chieh Wang, Ha Tran, Trevor R. Leonardo, Kristopher Nazor ,Ileana Slavin , Candace Lynch , Yingchun Li , Ronald Coleman , Irene Gallego Romero ,Gulsah Altun , David Reynolds , Stephen Dalton , Mana Parast , Jeanne F. Loring & Louise C. Laurent. Increased Risk of Genetic and Epigenetic Instability in Human Embryonic Stem Cells Associated with Specific Culture Conditions. PLOS ONE. 2015. DOI:10.1371/journal.pone.0118307. back to text
- Louise C. Laurent, Igor Ulitsky, Ileana Slavin, Ha Tran, Andrew Schork, Robert Morey, Candace Lynch, Julie V. Harness, Sunray Lee, Maria J. Barrero, Sherman Ku, Marina Martynova, Ruslan Semechkin, Vasiliy Galat, Joel Gottesfeld, Juan Carlos Izpisua Belmonte, Chuck Murry, Hans S. Keirstead, Hyun-Sook Park, Uli Schmidt, Andrew L. Laslett, Franz-Josef Muller, Caroline M. Nievergelt, Ron Shamir, Jeanne F. Loring. Dynamic Changes in the Copy Number of Pluripotency and Cell Proliferation Genes in Human ES and iPS Cells during Reprogramming and Time in Culture. Cell Stem Cell. 2011. Vol 8. 106-118. back to text
We are excited about the work these three scientists are doing and hope these studies will be vital steps toward beneficial cell therapies. We are privileged to be able to contribute our support.